Clustered regularly interspaced short palindromic repeats (CRISPR, pronounced crisper) are segments of prokaryotic DNA containing short repetitions of base ...
- Cas9 Cas9 (CRISPR associated protein 9) is an RNA-guided DNA...
- Gene drive Gene drive is the practice of "stimulating biased...
- CRISPR interference CRISPR interference (CRISPRi) is a genetic perturbation...
- Trans-activating crRNA In molecular biology, trans-activating crRNA (tracrRNA)...
CRISPR
From Wikipedia, the free encyclopedia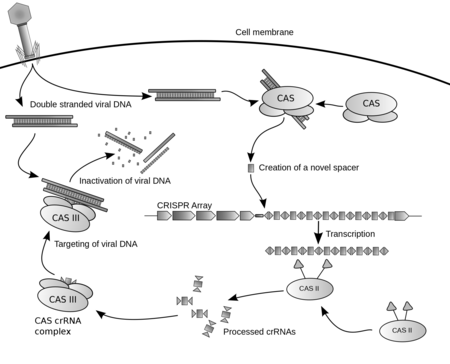
Clustered regularly interspaced short palindromic repeats (CRISPR, pronounced crisper[2]) are segments of prokaryotic DNA containing short repetitions of base sequences. Each repetition is followed by short segments of "spacer DNA" from previous exposures to a bacteriophage virus or plasmid.[3]Diagram of the CRISPR prokaryotic antiviral defense mechanism.[1]
The CRISPR/Cas system is a prokaryotic immune system that confers resistance to foreign genetic elements such as those present within plasmids and phages,[4][5][6] and provides a form of acquired immunity. The Cas protein(s) use the CRISPR spacers to recognize and cut these exogenous genetic elements in a manner analogous to RNA interference in eukaryotic organisms.[3] CRISPRs are found in approximately 40% of sequenced bacterial genomes and 90% of sequenced archaea.[7][note 1]
By delivering the Cas9 nuclease and appropriate guide RNAs into a cell, the cell's genome can be cut at a desired location, allowing existing genes to be removed and/or new ones added.[8][9][10] CRISPRs have been used in concert with specific endonuclease enzymes for genome editing and gene regulation in species throughout the tree of life.[11]
Cas9 was the first nuclease discovered, followed by Cpf1, which was discovered in the CRISPR/Cpf1 system of Francisella novicida.[12][13] Other such systems are thought to exist.[14]
CRISPR/C2c2 from the bacterium Leptotrichia shahii is RNA-guided CRISPR system that targets RNA rather than DNA, and can either cleave single-stranded RNA targets or knock them down.[15]
The CRISPR interference technique has many potential applications, including altering the germline of humans, animals, and food crops. The use of CRISPR for genome editing[16][17] was the AAAS's choice for breakthrough of the year in 2015.[18] Bioethical concerns have been expressed about the prospect of using this nascent biotechnology for editing the human germline.[19]
Contents
CRISPR 
Identifiers Organism Symbol ? History
Clustered DNA repeats with short unique sequences between the repeat units were first described in the bacterium Escherichia coli by Osaka University researcher Yoshizumi Ishino in 1987. The organization of the repeats was unusual because repeated sequences are typically arranged consecutively along the DNA. The function of the interrupted clustered repeats was not known at the time.[20][21] Repeats that were similarly interrupted were observed in an archaeal microbe by Spanish researcher Francisco Mojica in 1993.[22] He later named them short regularly spaced repeats (SRSR) after he found them in many other microbes.[23] SRSR was renamed CRISPR in 2002 after a suggestion from Mojica.[22][24] In the same year, a set of genes was found to be associated with CRISPR repeats and was named cas, or CRISPR-associated genes. The function of CRISPR was assumed to be related to the cas genes. Analysis of the protein sequences encoded by the cas genes suggested that one was a helicase that unwound DNA and that another was a nuclease that digested DNA.[24]

CRISPR was proposed to be responsible for adaptive immunity in microbes.[22] In 2005, three independent research groups showed that some CRISPR spacers are derived from phage DNA and extrachromosomal DNA such as plasmids.[25][26][27] In effect, the spacers are fragments of DNA gathered from viruses that previously tried to attack the cell. The source of the spacers was a sign that the CRISPR/cas system could have a role in adaptive immunity in bacteria.[1][28] All three studies proposing this idea were initially rejected by high-profile journals but were eventually published in other journals.[22]Simplified diagram of a CRISPR locus. The three major components of a CRISPR locus are shown: cas genes, a leader sequence, and a repeat-spacer array. Repeats are shown as gray boxes and spacers are colored bars. The arrangement of the three components is not always as shown.[1][3] In addition, several CRISPRs with similar sequences can be present in a single genome, only one of which is associated with cas genes.[7]
Although researchers came to an understanding that CRISPR-Cas was involved in microbial immunity, the mechanism of how the spacers conferred immunity was a mystery.[21] Koonin and colleagues proposed that spacers produce small RNA guides to target RNA transcribed from viral DNA, analogous to the RNA interference system used by eukaryotic cells.[21][29] Others hypothesized that CRISPR sequences directed Cas enzymes to degrade viral DNA.[21][27]
Experimental work by several groups revealed the basic mechanisms of CRISPR-Cas immunity. In 2007, Barrangou, Horvath, and other food industry scientists at Danisco provided the first experimental evidence that CRISPR was an adaptive immune system.[21] A CRISPR region in Streptococcus thermophilus acquired spacers from DNA of infecting bacteriophage. The researchers manipulated the resistance of S. thermophilus to phage by adding and deleting spacers whose sequence matched those found in the phages tested.[30][31] In 2008, Brouns and colleagues identified a complex of Cas protein that in E. coli cut the CRISPR RNA within the repeats into spacer-containing RNA molecules, which remained bound to the protein complex. In the same year, Marraffini and Sontheimer showed that a CRISPR sequence of Staphylococcus epidermidis targeted DNA and not RNA to prevent conjugation. This finding was at odds with the proposed RNA-interference-like mechanism of CRISPR-Cas immunity, although a CRISPR-Cas system that targets foreign RNA was later found in Pyrococcus furiosus.[21][30] A 2010 study provided direct evidence that CRISPR-Cas cuts both strands of phage and plasmid DNA in S. thermophilus.[32][33]
Cas9
Jennifer Doudna and Emmanuelle Charpentier studied a simpler CRISPR system from Streptococcus pyogenes that relies on the protein Cas9. The Cas9 endonuclease is a four-component system that includes two small RNA molecules.[34] In 2012, they re-engineered Cas9 endonuclease into a more manageable two-component system by fusing the two RNA molecules into a "single-guide RNA" that, when mixed with Cas9, could find and cut the DNA target specified by the guide RNA. By manipulating the nucleotide sequence of the guide RNA, the artificial Cas9 system could be programmed to target any sequence in DNA for cleavage.[35] This technological advance has fueled efforts to edit genomes with the re-engineered CRISPR-Cas9 system.[36]
Feng Zhang's and George Church's groups simultaneously described genome editing in human cell cultures using CRISPR-Cas9 systems for the first time.[21][37][38] It has since been used in a wide range of organisms, including baker's yeast (Saccharomyces cerevisiae),[39] zebrafish (D. rerio),[40] fruit flies (Drosophila melanogaster),[41] axolotl (A. mexicanum),[42] nematodes,(C. elegans),[43] plants,[44] mice,[45] monkeys[46] and human embryos.[47]
CRISPR has been modified to make programmable transcription factors that allow scientists to target and activate or silence specific genes.[48]
Libraries of tens of thousands of guide RNAs are available.[31]
Cpf1
In 2015, the nuclease Cpf1 was discovered in the CRISPR/Cpf1 system of the bacterium Francisella novicida.[12][13] Cpf1 showed several key differences to Cas9 including; causing a 'staggered' cut in double stranded DNA as opposed to the 'blunt' cut produced by Cas9, relying on a 'T rich' Protospacer adjacent motif (providing alternate targeting sites to Cas9) and requiring only a CRISPR RNA (crRNA) for successful targeting (with Cas9 requiring both crRNA and a transactivating crRNA (tracrRNA)).
Predecessors
In the early 2000s, researchers developed zinc finger nucleases, synthetic proteins whose DNA-binding domains enable them to create double-stranded breaks in DNA at specific points. In 2010, synthetic nucleases called TALENs provided an easier way to target a double-stranded break to a specific location on the DNA strand. Both zinc finger nucleases and TALENs require the creation of a custom protein for each targeted DNA sequence, which is a more difficult and time-consuming process than that for guide RNAs. CRISPRs are much easier to design because the process requires making only a short RNA sequence.[49]
Locus structure
Repeats and spacers
CRISPR repeats range in size from 24 to 48 base pairs.[50] They usually show some dyad symmetry, implying the formation of a secondary structure such as a hairpin, but are not truly palindromic.[51] Repeats are separated by spacers of similar length.[50] Some CRISPR spacer sequences exactly match sequences from plasmids and phages,[25][26][27] although some spacers match the prokaryote's genome (self-targeting spacers).[25][52] New spacers can be added rapidly as part of the immune response to phage infection.[53]
Cas genes and CRISPR subtypes
Small clusters of cas genes are often located next to CRISPR repeat-spacer arrays. Comparative genomics identified multiple cas genes; an initial analysis of 200 bacterial and archaeal genomes suggested as many as 45 cas gene families.[50]
The current classification groups CRISPR-Cas systems into two classes. Class 1 systems use a complex of multiple Cas proteins to degrade foreign nucleic acids. Class 2 systems use a single large Cas protein for the same purpose. Class 1 is divided into types I, III, and IV; class 2 is divided into types II and V. The five system types are divided into 16 subtypes. Each type and most subtypes are characterized by a "signature gene" found exclusively in the category. Classification is also based on the complement of cas genes that are present. Most CRISPR-Cas systems have a Cas1 protein. The phylogeny of Cas1 proteins generally agrees with the classification system.[54] Many organisms contain multiple CRISPR-Cas systems suggesting that they are compatible and may share components.[33][55] The sporadic distribution of the CRISPR/Cas subtypes suggests that the CRISPR/Cas system is subject to horizontal gene transfer during microbial evolution.
Signature genes and their putative functions for the major and minor CRISPR-cas types. Class Cas type Signature gene Function Reference 1 I Cas3 Single-stranded DNA nuclease (HD domain) and ATP-dependent helicase [56][57] IA Cas8a Subunit of the interference module. Important in targeting of invading DNA by recognizing the PAM sequence [58][59] IB Cas8b IC Cas8c ID Cas10d contains a domain homologous to the palm domain of nucleic acid polymerases and nucleotide cyclases [60][61] IE Cse1 IF Csy1 Not determined IU III Cas10 Homolog of Cas10d and Cse1 [61] IIIA Csm2 Not Determined IIIB Cmr5 Not Determined IIIC IIID IV Csf1 IVA IVB 2 II Cas9 Nucleases RuvC and HNH together produce DSBs, and separately can produce single-strand breaks. Ensures the acquisition of functional spacers during adaptation. [62][63] IIA Csn2 Ring-shaped DNA-binding protein. Involved in primed adaptation in Type II CRISPR system. [64] IIB Cas4 Not Determined IIC Characterized by the absence of either Csn2 or Cas4 [65] V Cpf1 Nuclease RuvC. Lacks HNH. Mechanism
CRISPR-Cas immunity is a natural process of bacteria and archaea. CRISPR-Cas prevents bacteriophage infection, conjugation, and natural transformation by degrading foreign nucleic acids that enter the cell.[30]
The stages of CRISPR immunity for each of the three major types of adaptive immunity. (1) Acquisition begins by recognition of invading DNA by Cas1 and Cas2 and cleavage of a protospacer. (2) The protospacer is ligated to the direct repeat adjacent to the leader sequence and (3) single strand extension repairs the CRISPR and duplicates the direct repeat. The crRNA processing and interference stages occur differently in each of the three major CRISPR systems. (4) The primary CRISPR transcript is cleaved by cas genes to produce crRNAs. (5) In type I systems Cas6e/Cas6f cleave at the junction of ssRNA and dsRNA formed by hairpin loops in the direct repeat. Type II systems use a trans-activating (tracr) RNA to form dsRNA, which is cleaved by Cas9 and RNaseIII. Type III systems use a Cas6 homolog that does not require hairpin loops in the direct repeat for cleavage. (6) In type II and type III systems secondary trimming is performed at either the 5’ or 3’ end to produce mature crRNAs. (7) Mature crRNAs associate with Cas proteins to form interference complexes. (8) In type I and type II systems, interactions between the protein and PAM sequence are required for degradation of invading DNA. Type III systems do not require a PAM for successful degradation and in type III-A systems basepairing occurs between the crRNA and mRNA rather than the DNA, targeted by type III-B systems.
Spacer acquisition
When a microbe is invaded by a virus, the first stage of the immune response is to capture viral DNA and insert it into a CRISPR locus in the form of a spacer. Cas1 and Cas2 are found in all three types of CRISPR-Cas immune systems, which indicates that they are involved in spacer acquisition. Mutation studies confirmed this hypothesis, showing that removal of cas1 or cas2 stopped spacer acquisition, without affecting CRISPR immune response.[58][66][67][68][69]
Multiple Cas1 proteins have been characterised and their structures resolved.[70][71][72] Cas1 proteins have diverse amino acid sequences. However, their crystal structures are similar and all purified Cas1 proteins are metal-dependent nucleases/integrases that bind to DNA in a sequence-independent manner.[33] Representative Cas2 proteins have been characterised and possess either ssRNA-[73] or dsDNA-[74][75] specific endoribonuclease activity.
In the I-E system of E. coli Cas1 and Cas2 form a complex where a Cas2 dimer bridges two Cas1 dimers.[76] In this complex Cas2 performs a non-enzymatic scaffolding role,[76] binding double-stranded fragments of invading DNA, while Cas1 binds the single-stranded flanks of the DNA and catalyses their integration into CRISPR arrays.[77][78][79]
Protospacer adjacent motifs
Main article: Protospacer adjacent motifBioinformatic analysis of regions of phage genomes that were excised as spacers (termed protospacers) revealed that they were not randomly selected but instead were found adjacent to short (3 – 5 bp) DNA sequences termed protospacer adjacent motifs (PAM). Analysis of CRISPR-Cas systems from the three major divisions showed PAMs to be important for type I and type II, but not type III systems during acquisition.[27][80][81][82][83][84] In type I and type II systems, protospacers are excised at positions adjacent to a PAM sequence, with the other end of the spacer cut using a ruler mechanism, thus maintaining the regularity of the spacer size in the CRISPR array.[85][86] The conservation of the PAM sequence differs between CRISPR-Cas systems and appears to be evolutionarily linked to Cas1 and the leader sequence.[84][87]
New spacers are added to a CRISPR array in a directional manner,[25] occurring preferentially,[53][80][81][88][89] but not exclusively, adjacent[83][86] to the leader sequence. Analysis of the type I-E system from E. coli demonstrated that the first direct repeat, adjacent to the leader sequence is copied, with the newly acquired spacer inserted between the first and second direct repeats.[68][85]
The PAM sequence appears to be important during spacer insertion in type I-E systems. That sequence contains a strongly conserved final nucleotide (nt) adjacent to the first nt of the protospacer. This nt becomes the final base in the first direct repeat.[69][90][91] This suggests that the spacer acquisition machinery generates single-stranded overhangs in the second-to-last position of the direct repeat and in the PAM during spacer insertion. However, not all CRISPR-Cas systems appear to share this mechanism as PAMs in other organisms do not show the same level of conservation in the final position.[87] It is likely that in those systems, a blunt end is generated at the very end of the direct repeat and the protospacer during acquisition.
Insertion variants
Analysis of Sulfolobus solfataricus CRISPRs revealed further complexities to the canonical model of spacer insertion, as one of its six CRISPR loci inserted new spacers randomly throughout its CRISPR array, as opposed to inserting closest to the leader sequence.[86]
Multiple CRISPRs contain many spacers to the same phage. The mechanism that causes this phenomenon was elucidated in the type I-E system of E. coli. A significant enhancement in spacer acquisition was detected where spacers already target the phage, even mismatches to the protospacer. This ‘priming’ requires the Cas proteins involved in both acquisition and interference to interact with each other. Newly acquired spacers that result from the priming mechanism are always found on the same strand as the spacer that caused the priming.[69][90][91] This observation led to the hypothesis that the acquisition machinery slides along the foreign DNA after priming to find a new protospacer.[91]
Biogenesis
CRISPR-RNA (crRNA), which later guides the Cas nuclease to the target during the interference step, must be generated from the CRISPR sequence. The crRNA is initially transcribed as part of a single long transcript encompassing much of the CRISPR array.[3] This transcript is then cleaved by Cas proteins to form crRNAs. The mechanism to produce crRNAs differs among CRISPR-Cas systems. In type I-E and type I-F systems, the proteins Cas6e and Cas6f respectively, recognise stem-loops[92][93][94] created by the pairing of identical repeats which flank the crRNA.[51] These Cas proteins cleave the longer transcript at the edge of the paired region, leaving a single crRNA along with a small remnant of the paired repeat region.
Type III systems also use Cas6, however their repeats do not produce stem-loops. Cleavage instead occurs by the longer transcript wrapping around the Cas6 to allow cleavage just upstream of the repeat sequence.[95][96][97]
Type II systems lack the Cas6 gene and instead utilize RNaseIII for cleavage. Functional type II systems encode an extra small RNA that is complementary to the repeat sequence, known as a trans-activating crRNA (tracrRNA).[66] Transcription of the tracrRNA and the primary CRISPR transcript results in base pairing and the formation of dsRNA at the repeat sequence, which is subsequently targeted by RNaseIII to produce crRNAs. Unlike the other two systems the crRNA does not contain the full spacer but instead is truncated at one end.[62]
CrRNAs associate with Cas proteins to form ribonucleotide complexes that recognize foreign nucleic acids. CrRNAs show no preference between the coding and non-coding strands, which is indicative of an RNA-guided DNA-targeting system.[6][32][58][69][98][99][100] The type I-E complex (commonly referred to as Cascade) requires five Cas proteins bound to a single crRNA.[101][102]
Interference
During the interference stage in type I systems the PAM sequence is recognized on the crRNA-complementary strand and is required along with crRNA annealing. In type I systems correct base pairing between the crRNA and the protospacer signals a conformational change in Cascade that recruits Cas3 for DNA degradation.
Type II systems rely on a single multifunctional protein, Cas9, for the interference step.[62] Cas9 requires both the crRNA and the tracrRNA to function and cleaves DNA using its dual HNH and RuvC/RNaseH-like endonuclease domains. Basepairing between the PAM and the phage genome is also required in type II systems, however the PAM is recognized on the same strand as the crRNA (the opposite strand to type I systems).
Type III systems, like type I require six or seven Cas proteins binding to crRNAs.[103][104] The type III systems analysed from S. solfataricus and P. furiosus both target the mRNA of phages rather than phage DNA genome,[55][104] which may make these systems uniquely capable of targeting RNA-based phage genomes.[33]
The mechanism for distinguishing self from foreign DNA during interference is built into the crRNAs and is therefore likely common to all three systems. Throughout the distinctive maturation process of each major type, all crRNAs contain a spacer sequence and some portion of the repeat at one or both ends. It is the partial repeat sequence that prevents the CRISPR-Cas system from targeting the chromosome as base pairing beyond the spacer sequence signals self and prevents DNA cleavage.[105] RNA-guided CRISPR enzymes are classified as type V restriction enzymes.
CRISPR associated protein 
crystal structure of a crispr-associated protein from Thermus thermophilusIdentifiers Symbol CRISPR_assoc Pfam PF08798 Pfam clan CL0362 InterPro IPR010179 CDD cd09727 [show]Available protein structures: CRISPR associated protein Cas2 
crystal structure of a hypothetical protein tt1823 from Thermus thermophilusIdentifiers Symbol CRISPR_Cas2 Pfam PF09827 InterPro IPR019199 CDD cd09638 [show]Available protein structures: CRISPR-associated protein Cse1 Identifiers Symbol CRISPR_Cse1 Pfam PF09481 InterPro IPR013381 CDD cd09729 [show]Available protein structures: CRISPR-associated protein Cse2 Identifiers Symbol CRISPR_Cse2 Pfam PF09485 InterPro IPR013382 CDD cd09670 [show]Available protein structures: Evolution and diversity
The basic model of CRISPR evolution is newly incorporated spacers driving phages to mutate their genomes to avoid the bacterial immune response, creating diversity in both the phage and host populations. To fight off a phage infection, the sequence of the CRISPR spacer must correspond perfectly to the sequence of the target phage gene. Phages can continue to infect their hosts given point mutations in the spacer.[105] Similar stringency is required in PAM or the bacterial strain remains phage sensitive.[81][105]
A study of 124 S. thermophilus strains showed that 26% of all spacers were unique and that different CRISPR loci showed different rates of new spacer acquisition.[80] Some CRISPR loci evolve more rapidly than others, which allowed the strains' phylogenetic relationships to be determined. A comparative genomic analysis showed that E. coli and S. enterica evolve much more slowly than S. thermophilus. The latter's strains that diverged 250 thousand years ago still contained the same spacer complement.[106]
Metagenomic analysis of two acid mine drainage biofilms showed that one of the analyzed CRISPRs contained extensive deletions and spacer additions versus the other biofilm, suggesting a higher phage activity/prevalence in one community than the other.[53] In the oral cavity, a temporal study determined that 7-22% of spacers were shared over 17 months within an individual while less than 2% were shared across individuals.[89]
From the same environment a single strain was tracked using PCR primers specific to its CRISPR system. Broad-level results of spacer presence/absence showed significant diversity. However, this CRISPR added 3 spacers over 17 months,[89] suggesting that even in an environment with significant CRISPR diversity some loci evolve slowly.
CRISPRs were analysed from the metagenomes produced for the human microbiome project.[107] Although most were body-site specific, some within a body site are widely shared among individuals. One of these loci originated from streptococcal species and contained ~15,000 spacers, 50% of which were unique. Similar to the targeted studies of the oral cavity, some showed little evolution over time.[107]
CRISPR evolution has been studied in chemostats using S. thermophilus to directly examine spacer acquisition rates. In one week, S. thermophilus strains acquired up to three spacers when challenged with a single phage.[108] During the same interval the phage developed single nucleotide polymorphisms that became fixed in the population, suggesting that targeting had prevented phage replication absent these mutations.[108] Another S. thermophilus experiments showed that phages can still infect and replicate in hosts that have only one targeting spacer. Yet another showed that sensitive hosts can exist in environments with high phage titres.[109] The chemostat and observational studies suggest many nuances to CRISPR and phage (co)evolution.
Identification
CRISPRs are widely distributed among bacteria and archaea[60] and show some sequence similarities.[51] Their most notable characteristic is their repeating spacers and direct repeats. This characteristic makes CRISPRs easily identifiable in long sequences of DNA, since the number of repeats decreases the likelihood of a false positive match. Three programs used for CRISPR repeat identification search for regularly interspaced repeats in long sequences: CRT,[110] PILER-CR[111] and CRISPRfinder.[112]
Analysis of CRISPRs in metagenomic data is more challenging, as CRISPR loci do not typically assemble, due to their repetitive nature or through strain variation, which confuses assembly algorithms. Where many reference genomes are available, polymerase chain reaction (PCR) can be used to amplify CRISPR arrays and analyse spacer content.[80][89][113][114][115] However, this approach yields information only for specifically targeted CRISPRs and for organisms with sufficient representation in public databases to design reliable PCR primers.
The alternative is to extract and reconstruct CRISPR arrays from shotgun metagenomic data. This is computationally more difficult, particularly with second generation sequencing technologies (e.g. 454, Illumina), as the short read lengths prevent more than two or three repeat units appearing in a single read. CRISPR identification in raw reads has been achieved using purely denovo identification[116] or by using direct repeat sequences in partially assembled CRISPR arrays from contigs (overlapping DNA segments that together represent a consensus region of DNA)[107] and direct repeat sequences from published genomes[117] as a hook for identifying direct repeats in individual reads.
Evolutionary significance
A bioinformatic study showed that CRISPRs are evolutionarily conserved and cluster into related types. Many show signs of a conserved secondary structure.[51]
Through the CRISPR/Cas mechanism, bacteria can acquire immunity to certain phages and thus halt further transmission of targeted phages. For this reason, Eugene Koonin has described CRISPR/Cas as a Lamarckian inheritance mechanism.[118] However, this has been disputed by a recent critic noting "We should remember [Lamarck] for the good he contributed to science, not for things that resemble his theory only superficially. Indeed, thinking of CRISPR and other phenomena as Lamarckian only obscures the simple and elegant way evolution really works.[119]
Analysis of CRISPR sequences revealed coevolution of host and viral genomes.[120] Cas9 proteins are highly enriched in pathogenic and commensal bacteria. CRISPR/Cas-mediated gene regulation may contribute to the regulation of endogenous bacterial genes, particularly during interaction with eukaryotic hosts. For example, Francisella novicida uses a unique, small, CRISPR/Cas-associated RNA (scaRNA) to repress an endogenous transcript encoding a bacterial lipoprotein that is critical for F. novicida to dampen host response and promote virulence.[121]
Use by phages
Another way for bacteria to defend against phage infection is by having chromosomal islands. A subtype of chromosomal islands called phage-inducible chromosomal island (PICI) is excised from a bacterial chromosome upon phage infection and can inhibit phage replication.[122] The mechanisms that induce PICI excision and how PICI inhibits phage replication are not well understood. One study showed that lytic ICP1 phage, which specifically targets Vibrio cholerae serogroup O1, has acquired a CRISPR/Cas system that targets a V. cholera PICI-like element. The system has 2 CRISPR loci and 9 Cas genes. It seems to be homologous to the 1-F system found in Yersinia pestis. Moreover, like the bacterial CRISPR/Cas system, ICP1 CRISPR/Cas can acquire new sequences, which allows phage and host to co-evolve.[123]
Applications
By the end of 2014 some 600 research papers had been published that mentioned CRISPR.[124] The technology had been used to functionally inactivate genes in human cell lines and cells, to study Candida albicans, to modify yeasts used to make biofuels and to genetically modify crop strains.[124]
Genome engineering
CRISPR/Cas9 genome editing is carried out with a Type II CRISPR system. When utilized for genome editing, this system includes Cas9, CRISPR RNA (crRNA), trans-activating crRNA (tracrRNA) along with an optional section of DNA repair template that is utilized in either Non-Homologous End Joining (NHEJ) or Homology Directed Repair (HDR).

Major components
CRISPR/Cas9 often employs a plasmid to transfect the target cells. The main components of this plasmid are displayed in the image and listed in the table. The crRNA needs to be designed for each application as this is the sequence that Cas9 uses to identify and directly bind to the cell's DNA. The crRNA must bind only where editing is desired. The repair template is designed for each application, as it must overlap with the sequences on either side of the cut and code for the insertion sequence.Component Function crRNA Contains the guide RNA that locates the correct section of host DNA along with a region that binds to tracrRNA (generally in a hairpin loop form) forming an active complex. tracrRNA Binds to crRNA and forms an active complex. sgRNA Single guide RNAs are a combined RNA consisting of a tracrRNA and at least one crRNA Cas9 Protein whose active form is able to modify DNA. Many variants exist with differing functions (i.e. single strand nicking, double strand break, DNA binding) due to Cas9's DNA site recognition function. Repair template DNA that guides the cellular repair process allowing insertion of a specific DNA sequence
Multiple crRNA's and the tracrRNA can be packaged together to form a single-guide RNA (sgRNA). This sgRNA can be joined together with the Cas9 gene and made into a plasmid in order to be transfected into cells (see image for overview).

overview of the transfection and DNA cleaving by CRISPR Cas9 (crRNA and tracrRNA are often joined as one strand of RNA when designing a plasmid)[127]Structure
CRISPR/Cas9 offers a high degree of fidelity and relatively simple construction. It depends on two factors for its specificity – the target sequence and the PAM. The target sequence is 20 bases long as part of each CRISPR locus in the crRNA array.[127] A typical crRNA array has multiple unique target sequences. Cas9 proteins select the correct location on the host's genome by utilizing the sequence to bond with base pairs on the host DNA. The sequence is not part of the Cas9 protein and as a result is customizable and can be independently synthesized.[128][129]
The PAM sequence on the host genome is recognized by the protein structure of Cas9 and generally cannot be easily modified to recognize a different sequence. However this is not too limiting as it is a short sequence and nonspecific (e.g. the SpCas9 PAM sequence is 5'-NGG-3' and in the human genome occurs roughly every 8 to 12 base pairs).[127]
Once these have been assembled into a plasmid and transfected into cells the Cas9 protein with help of the crRNA finds the correct sequence in the host cell's DNA and – depending on the Cas9 variant – creates a single or double strand break in the DNA.
Properly spaced single strand breaks in the host DNA can trigger homology directed repair, which is less error prone than non-homologous end joining that typically follows a double strand break. Providing a DNA repair template allows for the insertion of a specific DNA sequence at an exact location within the genome. The repair template should extend 40 to 90 base pairs beyond the Cas9 induced DNA break.[127] The goal is for the cell's HDR process to utilize the provided repair template and thereby incorporate the new sequence into the genome. Once incorporated, this new sequence is now part of the cell's genetic material and passes into its daughter cells.
Many online tools are available to aid in designing effective sgRNA sequences.[130]
Delivery
Scientists can use viral or non-viral system for delivery of the Cas9 and gRNA into target cells. Electroporation of DNA, RNA or ribonucleocomplexes is the most common and cheapest system. This technique was used to edit CXCR4 and PD-1, knocking in new sequences to replace specific genetic “letters” in these proteins. The group was then able to sort the cells, using cell surface markers, to help identify successfully edited cells.[131] Deep sequencing of a target site confirmed that knock-in genome modifications had occurred with up to ∼20% efficiency, which accounted for up to approximately one-third of total editing events.[132] However, hard-to-transfect cells (stem cells, neurons, hematopoietic cells, etc.) require more efficient delivery systems such as those based on lentivirus (LVs), adenovirus (AdV) and adeno-associated virus (AAV).
Editing
CRISPRs have been used to cut five[31] to 62 genes at once: pig cells have been engineered to inactivate all 62 Porcine Endogenous Retrovirus in the pig genome, which eliminated infection from the pig to human cells in culture.[133] CRISPR's low cost compared to alternatives is widely seen as revolutionary.[8][9]
Selective engineered redirection of the CRISPR/Cas system was first demonstrated in 2012 in:[134][135]
- Immunization of industrially important bacteria, including some used in food production and large-scale fermentation
- Cellular or organism RNA-guided genome engineering. Proof of concept studies demonstrated examples both in vitro[10][35][62] and in vivo[45][136][137]
- Bacterial strain discrimination by comparison of spacer sequences







Knockdown/activation
Main article: CRISPR interference
Using “dead” versions of Cas9 (dCas9)
eliminates CRISPR’s DNA-cutting ability, while preserving its ability
to target desirable sequences. Multiple groups added various regulatory
factors to dCas9s, enabling them to turn almost any gene on or off or
subtly adjust its level of activity.[138]
Like RNAi, CRISPR interference (CRISPRi) turns off genes in a
reversible fashion by targeting, but not cutting a site. The targeted
site is methylated, epigenetically
modifying the gene. This modification inhibits transcription. Cas9 is
an effective way of targeting and silencing specific genes at the DNA
level.[139]
In bacteria, the presence of Cas9 alone is enough to block
transcription. For mammalian applications, a section of protein is
added. Its guide RNA targets regulatory DNA sequences called promoters that immediately precede the target gene.[31]Cas9 was used to carry synthetic transcription factors that activated specific human genes. The technique achieved a strong effect by targeting multiple CRISPR constructs to slightly different locations on the gene's promoter.[31]
RNA editing
In 2016 researchers demonstrated that CRISPR from an ordinary mouth bacterium could be used to edit RNA. The researchers searched databases containing hundreds of millions of genetic sequences for those that resembled Crispr genes. With Zhang they considered the fusobacteria Leptotrichia shahii. It had a group of genes that resembled CRISPR genes, but with important differences. When the researchers equipped other bacteria with these genes, which they called C2c2, they found that the organisms gained a novel defense.[140]Many viruses encode their genetic information in RNA rather than DNA that they repurpose to make new viruses. HIV and poliovirus are such viruses. Bacteria with C2c2 make molecules that can dismember RNA, destroying the virus. Tailoring these genes opened any RNA molecule to editing.[140]
Disease models
CRISPR simplifies creation of animals for research that mimic disease or show what happens when a gene is knocked down or mutated. CRISPR may be used at the germline level to create animals where the gene is changed everywhere, or it may be targeted at non-germline cells.[141][142][143]CRISPR can be utilized to create human cellular models of disease. For instance, CRISPR was applied to human pluripotent stem cells to introduce targeted mutations in genes relevant to polycystic kidney disease (PKD) and focal segmental glomerulosclerosis (FSG).[144] These CRISPR-modified pluripotent stem cells were subsequently grown into human kidney organoids that exhibited disease-specific phenotypes. Kidney organoids from stem cells with PKD populations formed large, translucent cyst structures from kidney tubules. Kidney organoids with mutations in a gene linked to FSG developed junctional defects between podocytes, the filtering cells affected in that disease. Importantly, these disease phenotypes were absent in control organoids of identical genetic background, but lacking the CRISPR modifications.[144]
A similar approach has been taken to model long QT syndrome in cardiomyocytes derived from pluripotent stem cells.[145] These CRISPR-generated cellular models, with isogenic controls, provide a new way to study human diseases and test drugs.
Gene drive
In 2003 evolutionary biologist Austin Burt envisioned attaching a gene that coded for a desired trait to “selfish” DNA elements that could copy themselves from one chromosome position to another. That would bias daughter cells to inherit it, quickly spreading it throughout a population. In 2015 a U.S. team used CRISPR to create a “mutagenic chain reaction” that drove a pigmentation trait in lab-grown Drosophila to the next generation with 97% efficiency. With another research group they created a gene drive in mosquitoes that spread genes that prevented the insects from harboring malaria parasites. Only weeks later, the team reported a second drive with genes that rendered female mosquitoes infertile and could quickly wipe out a population. The work was done in the lab, stimulating debates over the desirability of field testing.[138]Biomedicine
CRISPR/Cas-based "RNA-guided nucleases" can be used to target virulence factors, genes encoding antibiotic resistance, and other medically relevant sequences of interest. This technology thus represents a novel form of antimicrobial therapy and a strategy by which to manipulate bacterial populations.[146] Some of the affected genes are tied to human diseases, including those involved in muscle differentiation, cancer, inflammation and fetal hemoglobin.[31]Research suggests that CRISPR is an effective way to limit replication of multiple herpesviruses. It was even able to eradicate viral DNA in the case of Epstein-Barr virus (EBV). Anti-herpesvirus CRISPRs have several promising applications, such as removing cancer-causing EBV from tumor cells, helping rid donated organs for immunocompromised patients of viral invaders, or preventing cold sore outbreaks and recurrent eye infections by blocking HSV-1 reactivation. As of August 2016, these await testing in animal models or humans.[147]
Clinical researchers are applying it to develop tissue-based treatments for cancer and other diseases.[138]
CRISPR may revive the concept of transplanting animal organs into people. Retroviruses present in animal genomes could harm transplant recipients. In 2015 a team eliminated 62 copies of a retrovirus’s DNA from the pig genome.[138]
It may also have applications in tissue engineering and regenerative medicine, such as by creating human blood vessels that lack expression of MHC class II proteins, which often cause transplant rejection.[148]
Gene function
In 2015, multiple studies attempted to systematically disable each individual human gene, in an attempt to identify which genes were essential to human biology. Between 1,600 and 1,800 genes passed this test—of the 20,000 or so known human genes. Such genes are more strongly activated, and unlikely to carry disabling mutations. They are more likely to have indispensable counterparts in other species. They build proteins that unite to form larger collaborative complexes. The studies also catalogued the essential genes in four cancer-cell lines and identified genes that are expendable in healthy cells, but crucial in specific tumor types and drugs that could target these rogue genes.[149]The specific functions of some 18 percent of the essential genes are unidentified. In one 2015 targeting experiment, disabling individual genes in groups of cells attempted to identify those involved in resistance to a melanoma drug. Each such gene manipulation is itself a separate "drug", potentially opening the entire genome to CRISPR-based regulation.[138]
In vitro genetic depletion
Unenriched sequencing libraries often have abundant undesired sequences. Cas9 can specifically deplete the undesired sequences with double strand breakage with up to 99% efficiency and without significant off-target effects as seen with restriction enzymes. Treatment with Cas9 can deplete abundant rRNA while increasing pathogen sensitivity in RNA-seq libraries.[150]Patents and commercialization
As of December 2014, patent rights to CRISPR were contested. Several companies had formed to develop related drugs and research tools.[151] As companies ramp up financing, doubts as to whether or not CRISPR can be quickly monetized were raised.[152]As of November 2013, SAGE Labs (now part of Horizon Discovery group) had exclusive rights from one of those companies to produce and sell genetically engineered rats and non-exclusive rights for mouse and rabbit models.[153] By 2015, Thermo Fisher Scientific had licensed intellectual property from ToolGen to develop CRISPR reagent kits.[154]
Society and culture
Human germline modification
At least four labs in the US, labs in China and the UK, and by a US biotechnology company called Ovascience announced plans or ongoing research to apply CRISPR to human embryos.[155] Scientists, including a CRISPR co-inventor, urged a worldwide moratorium on applying CRISPR to the human germline, especially for clinical use. They said "scientists should avoid even attempting, in lax jurisdictions, germline genome modification for clinical application in humans" until the full implications "are discussed among scientific and governmental organizations".[47][156] These scientists support basic research on CRISPR and do not see CRISPR as developed enough for any clinical use in making heritable changes to people.[157]In April 2015, Chinese scientists reported results of an attempt to alter the DNA of non-viable human embryos using CRISPR to correct a mutation that causes beta thalassemia, a lethal heritable disorder.[158][159] The study had previously been rejected by both Nature and Science in part because of ethical concerns; the journals had no comment.[160] The experiments resulted in changing only some genes, and had off-target effects on other genes. The researchers stated that CRISPR is not ready for clinical application in reproductive medicine."[160] In April 2016 Chinese scientists were reported to have made a second unsuccessful attempt to alter the DNA of non-viable human embryos using CRISPR - this time to alter the CCR5 gene to make the embryo HIV resistant.[161]
In December 2015, the International Summit on Human Gene Editing took place in Washington under the guidance of David Baltimore. Members of national scientific academies of America, Britain and China discussed the ethics of germline modification. They agreed to support basic and clinical research under appropriate legal and ethical guidelines. A specific distinction was made between clinical use in somatic cells, where the effects of edits are limited to a single individual, versus germline cells, where genome changes could be inherited by future generations. This could have unintended and far-reaching and potentially damaging consequences for human evolution, genetically (e.g. gene/environment interactions) and culturally (e.g. Social Darwinism). Altering of gametocytes and embryos to generate inheritable changes in humans was thus claimed irresponsible. In addition, they agreed to initiate an international forum to address such concerns and harmonize regulations countries.[162]
In February 2016, British scientists were given permission by regulators to genetically modify human embryos by using CRISPR-Cas9 and related techniques. The embryos were to be destroyed after seven days.[163][164]
Recognition
In 2012 and 2013, CRISPR was a runner-up in Science Magazine's Breakthrough of the Year award. In 2015, it was the winner of that award.[138] CRISPR was named as one of MIT Technology Review's 10 breakthroughs technologies in 2014 and 2016.[165][166]Alternative cutters










See also
Notes
- 71/79 Archaea, 463/1008 Bacteria CRISPRdb, Date: 19.6.2010 Archived May 16, 2015, at the Wayback Machine.
References
- Larson C, Schaffer A (2014). "Genome Editing/ 10 Breakthrough Technologies 2014". Massachusetts Institute of Technology. Retrieved 18 March 2016.
Further reading
- CRISPR-Cas: A Laboratory Manual Edited by Jennifer Doudna, University of California, Berkeley; Prashant Mali, University of California, San Diego
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J (August 2016). "Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems". Science. 353 (6299). doi:10.1126/science.aad5147.
- Sander JD, Joung JK (April 2014). "CRISPR-Cas systems for editing, regulating and targeting genomes". Nature Biotechnology. 32 (4): 347–55. doi:10.1038/nbt.2842. PMID 24584096.
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (January 2016). "Rationally engineered Cas9 nucleases with improved specificity". Science. 351 (6268): 84–8. doi:10.1126/science.aad5227.
- Terns RM, Terns MP (March 2014). "CRISPR-based technologies: prokaryotic defense weapons repurposed". Trends in Genetics. 30 (3): 111–8. doi:10.1016/j.tig.2014.01.003. PMID 24555991.
- Westra ER, Buckling A, Fineran PC (May 2014). "CRISPR-Cas systems: beyond adaptive immunity". Nature Reviews. Microbiology. 12 (5): 317–26. doi:10.1038/nrmicro3241. PMID 24704746.
- Andersson AF, Banfield JF (May 2008). "Virus population dynamics and acquired virus resistance in natural microbial communities". Science. 320 (5879): 1047–50. Bibcode:2008Sci...320.1047A. doi:10.1126/science.1157358. PMID 18497291.
- Hale C, Kleppe K, Terns RM, Terns MP (December 2008). "Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus". RNA. 14 (12): 2572–9. doi:10.1261/rna.1246808. PMC 2590957
 . PMID 18971321.
. PMID 18971321. - van der Ploeg JR (June 2009). "Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages". Microbiology. 155 (Pt 6): 1966–76. doi:10.1099/mic.0.027508-0. PMID 19383692.
- van der Oost J, Brouns SJ (November 2009). "RNAi: prokaryotes get in on the act". Cell. 139 (5): 863–5. doi:10.1016/j.cell.2009.11.018. PMID 19945373.
- Karginov FV, Hannon GJ (January 2010). "The CRISPR system: small RNA-guided defense in bacteria and archaea". Molecular Cell. 37 (1): 7–19. doi:10.1016/j.molcel.2009.12.033. PMC 2819186. PMID 20129051.
- Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R (March 2010). "Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS". Molecular Microbiology. 75 (6): 1495–512. doi:10.1111/j.1365-2958.2010.07073.x. PMID 20132443.
- Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJ (May 2010). "Diversity of CRISPR loci in Escherichia coli". Microbiology. 156 (Pt 5): 1351–61. doi:10.1099/mic.0.036046-0. PMID 20133361.
- Deveau H, Garneau JE, Moineau S (2010). "CRISPR/Cas system and its role in phage-bacteria interactions". Annual Review of Microbiology. 64: 475–93. doi:10.1146/annurev.micro.112408.134123. PMID 20528693.
- Koonin EV, Makarova KS (December 2009). "CRISPR-Cas: an adaptive immunity system in prokaryotes". F1000 Biology Reports. 1: 95. doi:10.3410/B1-95. PMC 2884157. PMID 20556198.
- "The age of the red pen". The Economist. August 22, 2015. ISSN 0013-0613. Retrieved 2015-08-25.
- Lander ES (January 2016). "The Heroes of CRISPR". Cell. 164 (1-2): 18–28. doi:10.1016/j.cell.2015.12.041. PMID 26771483.

Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA (September 2013). "Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis". Proceedings of the National Academy of Sciences of the United States of America. 110 (39): 15644–9. Bibcode:2013PNAS..11015644H. doi:10.1073/pnas.1313587110.
CRISPR Patents Spark Fight to Control Genome Editing
The biologists writing in Science support continuing laboratory research with the technique, and few if any scientists believe it is ready for clinical use.
No comments:
Post a Comment