Sintering
From Wikipedia, the free encyclopedia
Clinker nodules produced by sintering
Sintering is the process of compacting and forming a solid mass of material by heat
[1] and/or pressure
[2] without melting it to the point of liquefaction.
Sintering happens naturally in mineral deposits or as a manufacturing
process used with metals, ceramics, plastics, and other materials. The
atoms in the materials diffuse across the boundaries of the particles,
fusing the particles together and creating one solid piece. Because the
sintering temperature does not have to reach the melting point of the
material, sintering is often chosen as the shaping process for materials
with extremely high melting points such as
tungsten and
molybdenum. The study of sintering in metallurgy powder-related processes is known as
powder metallurgy.
An example of sintering can be observed when ice cubes in a glass of
water adhere to each other, which is driven by the temperature
difference between the water and the ice.
[citation needed]
Examples of pressure-driven sintering are the compacting of snowfall to
a glacier, or the forming of a hard snowball by pressing loose snow
together.
The word "sinter" comes from the
Middle High German sinter, a
cognate of English "
cinder".
General sintering
Sintering is effective when the process reduces the
porosity and enhances properties such as strength,
electrical conductivity,
translucency and
thermal conductivity;
yet, in other cases, it may be useful to increase its strength but keep
its gas absorbency constant as in filters or catalysts.
[citation needed]
During the firing process, atomic diffusion drives powder surface
elimination in different stages, starting from the formation of necks
between powders to final elimination of small pores at the end of the
process.
The driving force for densification is the change in free energy from
the decrease in surface area and lowering of the surface free energy by
the replacement of solid-vapor interfaces. It forms new but
lower-energy solid-solid interfaces with a total decrease in free energy
occurring on sintering 1-micrometre particles a 1 cal/g decrease. On a
microscopic scale, material transfer is affected by the change in
pressure and differences in free energy across the curved surface. If
the size of the particle is small (and its curvature is high), these
effects become very large in magnitude. The change in energy is much
higher when the radius of curvature is less than a few micrometres,
which is one of the main reasons why much ceramic technology is based on
the use of fine-particle materials.
[3]
For properties such as strength and conductivity, the bond area in
relation to the particle size is the determining factor. The variables
that can be controlled for any given material are the temperature and
the initial grain size, because the vapor pressure depends upon
temperature. Through time, the particle radius and the vapor pressure
are proportional to (p
0)
2/3 and to (p
0)
1/3, respectively.
[3]
The source of power for solid-state processes is the change in free
or chemical potential energy between the neck and the surface of the
particle. This energy creates a transfer of material through the fastest
means possible; if transfer were to take place from the particle volume
or the grain boundary between particles, then there would be particle
reduction and pore destruction. The pore elimination occurs faster for a
trial with many pores of uniform size and higher porosity where the
boundary diffusion distance is smaller. For the latter portions of the
process, boundary and lattice diffusion from the boundary become
important.
[3]
Control of temperature is very important to the sintering process,
since grain-boundary diffusion and volume diffusion rely heavily upon
temperature, the size and distribution of particles of the material, the
materials composition, and often the sintering environment to be
controlled.
[3]
Ceramic sintering
Sintering is part of the firing process used in the manufacture of
pottery and other ceramic objects. These objects are made from substances such as
glass,
alumina,
zirconia,
silica,
magnesia,
lime,
beryllium oxide, and
ferric oxide. Some ceramic raw materials have a lower
affinity for water and a lower
plasticity index than
clay,
requiring organic additives in the stages before sintering. The general
procedure of creating ceramic objects via sintering of powders
includes:
- Mixing water, binder, deflocculant, and unfired ceramic powder to form a slurry;
- Spray-drying the slurry;
- Putting the spray dried powder into a mold and pressing it to form a green body (an unsintered ceramic item);
- Heating the green body at low temperature to burn off the binder;
- Sintering at a high temperature to fuse the ceramic particles together.
All the characteristic temperatures associated with phase
transformation, glass transitions, and melting points, occurring during a
sinterisation cycle of a particular ceramics formulation (i.e., tails
and frits) can be easily obtained by observing the expansion-temperature
curves during
optical dilatometer
thermal analysis. In fact, sinterisation is associated with a
remarkable shrinkage of the material because glass phases flow once
their transition temperature is reached, and start consolidating the
powdery structure and considerably reducing the porosity of the
material.
There are two types of sintering: with pressure (also known as
hot pressing),
and without pressure. Pressureless sintering is possible with graded
metal-ceramic composites, with a nanoparticle sintering aid and bulk
molding technology. A variant used for 3D shapes is called
hot isostatic pressing.
To allow efficient stacking of product in the furnace during
sintering and prevent parts sticking together, many manufacturers
separate ware using ceramic powder separator sheets. These sheets are
available in various materials such as alumina, zirconia and magnesia.
They are additionally categorized by fine, medium and coarse particle
sizes. By matching the material and particle size to the ware being
sintered, surface damage and contamination can be reduced while
maximizing furnace loading.
Sintering of metallic powders
Most, if not all, metals can be sintered. This applies especially to
pure metals produced in vacuum which suffer no surface contamination.
Sintering under atmospheric pressure requires the use of a protective
gas, quite often
endothermic gas.
[4] Sintering, with subsequent reworking, can produce a great range of material properties. Changes in density,
alloying, or heat treatments can alter the physical characteristics of various products. For instance, the
Young's Modulus En of sintered
iron
powders remains insensitive to sintering time, alloying, or particle
size in the original powder, but depends upon the density of the final
product:

where
D is the density,
E is
Young's modulus and
d is the maximum density of iron.
Sintering is static when a metal powder under certain external
conditions may exhibit coalescence, and yet reverts to its normal
behavior when such conditions are removed. In most cases, the density of
a collection of grains increases as material flows into voids, causing a
decrease in overall volume. Mass movements that occur during sintering
consist of the reduction of total porosity by repacking, followed by
material transport due to
evaporation and
condensation from
diffusion.
In the final stages, metal atoms move along crystal boundaries to the
walls of internal pores, redistributing mass from the internal bulk of
the object and smoothing pore walls.
Surface tension is the driving force for this movement.
A special form of sintering (which is still considered part of powder
metallurgy) is liquid-state sintering in which at least one but not all
elements are in a liquid state. Liquid-state sintering is required for
making
cemented carbide or
tungsten carbide.
Sintered
bronze in particular is frequently used as a material for
bearings,
since its porosity allows lubricants to flow through it or remain
captured within it. Sintered copper may be used as a wicking structure
in certain types of
heat pipe construction, where the porosity allows a liquid agent to move through the porous material via
capillary action. For materials that have high melting points such as
molybdenum,
tungsten,
rhenium,
tantalum,
osmium and
carbon,
sintering is one of the few viable manufacturing processes. In these
cases, very low porosity is desirable and can often be achieved.
Sintered metal powder is used to make
frangible shotgun shells called
breaching rounds,
as used by military and SWAT teams to quickly force entry into a locked
room. These shotgun shells are designed to destroy door deadbolts,
locks and hinges without risking lives by ricocheting or by flying on at
lethal speed through the door. They work by destroying the object they
hit and then dispersing into a relatively harmless powder.
Sintered bronze and stainless steel are used as filter materials in
applications requiring high temperature resistance while retaining the
ability to regenerate the filter element. For example, sintered
stainless steel elements are employed for filtering steam in food and
pharmaceutical applications, and sintered bronze in aircraft hydraulic
systems.
Sintering of powders containing precious metals such as
silver and
gold is used to make small jewelry items.
Advantages
Particular advantages of the powder technology include:
- Very high levels of purity and uniformity in starting materials
- Preservation of purity, due to the simpler subsequent fabrication process (fewer steps) that it makes possible
- Stabilization of the details of repetitive operations, by control of grain size during the input stages
- Absence of binding contact between segregated powder particles – or
"inclusions" (called stringering) – as often occurs in melting processes
- No deformation needed to produce directional elongation of grains
- Capability to produce materials of controlled, uniform porosity.
- Capability to produce nearly net-shaped objects.
- Capability to produce materials which cannot be produced by any other technology.
- Capability to fabricate high-strength material like turbine blades.
- After sintering the mechanical strength to handling becomes higher.
- Very Sexy
The literature contains many references on sintering dissimilar
materials to produce solid/solid-phase compounds or solid/melt mixtures
at the processing stage. Almost any substance can be obtained in powder
form, through either chemical, mechanical or physical processes, so
basically any material can be obtained through sintering. When pure
elements are sintered, the leftover powder is still pure, so it can be
recycled.
Disadvantages
Particular disadvantages of the powder technology include:
- 100% sintered (iron ore) can not be charged in the blast furnace.
- By sintering one cannot create uniform sizes.
- Micro- and nano-structures produced before sintering are often destroyed.
Plastics sintering
Plastic materials are formed by sintering for applications that
require materials of specific porosity. Sintered plastic porous
components are used in filtration and to control fluid and gas flows.
Sintered plastics are used in applications requiring wicking properties,
such as marking pen nibs. Sintered
ultra high molecular weight polyethylene materials are used as
ski and
snowboard
base materials. The porous texture allows wax to be retained within the
structure of the base material, thus providing a more durable wax
coating.
Liquid phase sintering
For materials which are difficult to sinter, a process called liquid
phase sintering is commonly used. Materials for which liquid phase
sintering is common are
Si3N4,
WC,
SiC,
and more. Liquid phase sintering is the process of adding an additive
to the powder which will melt before the matrix phase. The process of
liquid phase sintering has three stages:
- Rearrangement – As the liquid melts capillary action will
pull the liquid into pores and also cause grains to rearrange into a
more favorable packing arrangement.
- Solution-Precipitation – In areas where capillary pressures
are high (particles are close together) atoms will preferentially go
into solution and then precipitate in areas of lower chemical potential
where particles are not close or in contact. This is called "contact flattening". This densifies the system in a way similar to grain boundary diffusion in solid state sintering. Ostwald ripening
will also occur where smaller particles will go into solution
preferentially and precipitate on larger particles leading to
densification.
- Final Densification – densification of solid skeletal network, liquid movement from efficiently packed regions into pores.
For liquid phase sintering to be practical the major phase should be
at least slightly soluble in the liquid phase and the additive should
melt before any major sintering of the solid particulate network occurs,
otherwise rearrangement of grains will not occur. Liquid phase
sintering was successfully applied to improve
grain growth of thin semiconductor layers from
nanoparticle precursor films.
[5]
Electric current assisted sintering
These techniques employ electric currents to drive or enhance sintering.
[6] English engineer A. G. Bloxam registered in 1906 the first
patent on sintering powders using
direct current in
vacuum. The primary purpose of his inventions was the industrial scale production of filaments for
incandescent lamps by compacting
tungsten or
molybdenum particles. The applied current was particularly effective in reducing surface
oxides that increased the
emissivity of the filaments.
[7]
In 1913, Weintraub and Rush patented a modified sintering method which combined electric current with
pressure. The benefits of this method were proved for the sintering of
refractory metals as well as conductive
carbide or
nitride powders. The starting
boron–
carbon or
silicon–carbon powders were placed in an
electrically insulating tube and compressed by two rods which also served as
electrodes for the current. The estimated sintering temperature was 2000 °C.
[7]
In the United States, sintering was first patented by Duval d’Adrian
in 1922. His three-step process aimed at producing heat-resistant blocks
from such oxide materials as
zirconia,
thoria or
tantalia. The steps were: (i)
molding the powder; (ii)
annealing it at about 2500 °C to make it conducting; (iii) applying current-pressure sintering as in the method by Weintraub and Rush.
[7]
Sintering that uses an
arc produced via a
capacitance
discharge to eliminate oxides before direct current heating, was
patented by G. F. Taylor in 1932. This originated sintering methods
employing pulsed or
alternating current,
eventually superimposed to a direct current. Those techniques have been
developed over many decades and summarized in more than 640 patents.
[7]
Of these technologies the most well known is resistance sintering (also called
hot pressing) and
spark plasma sintering, while
Electro Sinter Forging is the latest advancement in this field.
Spark plasma sintering
In
spark plasma sintering
(SPS), external pressure and an electric field are applied
simultaneously to enhance the densification of the metallic/ceramic
powder compacts. This densification uses lower temperatures and shorter
amount of time than typical sintering.
[8]
For a number of years, it was speculated that the existence of sparks
or plasma between particles could aid sintering; however, Hulbert and
coworkers systematically proved that the electric parameters used during
spark plasma sintering make it (highly) unlikely.
[9]
In light of this, the name "spark plasma sintering" has been rendered
obsolete. Terms such as "Field Assisted Sintering Technique" (FAST),
"Electric Field Assisted Sintering" (EFAS), and Direct Current Sintering
(DCS) have been implemented by the sintering community.
[10]
Using a DC pulse as the electric current, spark plasma, spark impact
pressure, joule heating, and an electrical field diffusion effect would
be created.
[11]
Electro Sinter Forging
Electro Sinter Forging is an electric current assisted sintering (ecas) technology originated from
Capacitor discharge sintering. It is used for the production of diamond metal matrix composites and under evaluation for the production of hard metals,
[12] nitinol
[13]
and other metals and intermetallics. It is characterized by a very low
sintering time allowing machines to sinter at the same speed as a
compaction press.
Pressureless sintering
Pressureless sintering is the sintering of a powder compact
(sometimes at very high temperatures, depending on the powder) without
applied pressure. This avoids density variations in the final component,
which occurs with more traditional hot pressing methods.
[14]
The powder compact (if a ceramic) can be created by
slip casting,
injection moulding, and
cold isostatic pressing. After pre-sintering, the final green compact can be machined to its final shape before sintered.
Three different heating schedules can be performed with pressureless
sintering: constant-rate of heating (CRH), rate-controlled sintering
(RCS), and two-step sintering (TSS). The microstructure and grain size
of the ceramics may vary depending on the material and method used.
[14]
Constant-rate of heating (CRH), also known as temperature-controlled
sintering, consists of heating the green compact at a constant rate up
to the sintering temperature.
[15]
Experiments with zirconia have been performed to optimize the sintering
temperature and sintering rate for CRH method. Results showed that the
grain sizes were identical when the samples were sintered to the same
density, proving that grain size is a function of specimen density
rather than CRH temperature mode.
In rate-controlled sintering (RCS), the densification rate in the open-porosity phase is lower than in the CRH method.
[15] By definition, the relative density, ρ
rel,
in open-porosity phase is lower than 90%. Although this should prevent
separation of pores from grain boundaries, it has been proven
statistically that RCS did not produce smaller grain sizes than CRH for
alumina, zirconia, and ceria samples.
[14]
Two-step sintering (TSS) uses two different sintering temperatures.
The first sintering temperature should guarantee a relative density
higher than 75% of theoretical sample density. This will remove
supercritical pores from the body. The sample will then be cooled down
and held at the second sintering temperature until densification is
completed. Grains of cubic zirconia and cubic strontium titanate were
significantly refined by TSS compared to CRH. However, the grain size
changes in other ceramic materials, like tetragonal zirconia and
hexagonal alumina, were not statistically significant.
[14]
Densification, vitrification and grain growth
Sintering in practice is the control of both densification and
grain growth.
Densification is the act of reducing porosity in a sample thereby
making it more dense. Grain growth is the process of grain boundary
motion and
Ostwald ripening to increase the average grain size. Many properties (
mechanical strength, electrical breakdown strength, etc.) benefit from both a high relative
density
and a small grain size. Therefore, being able to control these
properties during processing is of high technical importance. Since
densification of powders requires high temperatures, grain growth
naturally occurs during sintering. Reduction of this process is key for
many engineering ceramics.
For densification to occur at a quick pace it is essential to have
(1) an amount of liquid phase that is large in size, (2) a near complete
solubility of the solid in the liquid, and (3) wetting of the solid by
the liquid. The power behind the densification is derived from the
capillary pressure of the liquid phase located between the fine solid
particles. When the liquid phase wets the solid particles, each space
between the particles becomes a capillary in which a substantial
capillary pressure is developed. For submicrometre particle sizes,
capillaries with diameters in the range of 0.1 to 1 micrometres develop
pressures in the range of 175 pounds per square inch (1,210 kPa) to
1,750 pounds per square inch (12,100 kPa) for silicate liquids and in
the range of 975 pounds per square inch (6,720 kPa) to 9,750 pounds per
square inch (67,200 kPa) for a metal such as liquid cobalt.
[3]
Densification requires constant
capillary pressure
where just solution-precipitation material transfer would not produce
densification. For further densification, additional particle movement
while the particle undergoes grain-growth and grain-shape changes
occurs. Shrinkage would result when the liquid slips between particles
and increase pressure at points of contact causing the material to move
away from the contact areas forcing particle centers to draw near each
other.
[3]
The sintering of liquid-phase materials involves a fine-grained solid
phase to create the needed capillary pressures proportional to its
diameter and the liquid concentration must also create the required
capillary pressure within range, else the process ceases. The
vitrification rate is dependent upon the pore size, the viscosity and
amount of liquid phase present leading to the viscosity of the overall
composition, and the surface tension. Temperature dependence for
densification controls the process because at higher temperatures
viscosity decreases and increases liquid content. Therefore, when
changes to the composition and processing are made, it will affect the
vitrification process.
[3]
Sintering mechanisms
Sintering occurs by diffusion of atoms through the microstructure.
This diffusion is caused by a gradient of chemical potential – atoms
move from an area of higher chemical potential to an area of lower
chemical potential. The different paths the atoms take to get from one
spot to another are the sintering mechanisms. The six common mechanisms
are:
- Surface diffusion – Diffusion of atoms along the surface of a particle
- Vapor transport – Evaporation of atoms which condense on a different surface
- Lattice diffusion from surface – atoms from surface diffuse through lattice
- Lattice diffusion from grain boundary – atom from grain boundary diffuses through lattice
- Grain boundary diffusion – atoms diffuse along grain boundary
- Plastic deformation – dislocation motion causes flow of matter
Also one must distinguish between densifying and non-densifying mechanisms. 1–3 above are non-densifying
[citation needed]
– they take atoms from the surface and rearrange them onto another
surface or part of the same surface. These mechanisms simply rearrange
matter inside of porosity and do not cause pores to shrink. Mechanisms
4–6 are densifying mechanisms
[citation needed] – atoms are moved from the bulk to the surface of pores thereby eliminating porosity and increasing the density of the sample.
Grain growth
Main article:
Grain growth
A
grain boundary(GB) is the transition area or interface between adjacent
crystallites (or
grains) of the same chemical and
lattice composition, not to be confused with a
phase boundary.
The adjacent grains do not have the same orientation of the lattice
thus giving the atoms in GB shifted positions relative to the lattice in
the
crystals.
Due to the shifted positioning of the atoms in the GB they have a
higher energy state when compared with the atoms in the crystal lattice
of the grains. It is this imperfection that makes it possible to
selectively etch the GBs when one wants the microstructure visible.
[16] Striving to minimize its energy leads to the coarsening of the
microstructure to reach a metastable state within the specimen. This involves minimizing its GB area and changing its
topological structure to minimize its energy. This grain growth can either be
normal or abnormal, a normal grain growth is characterized by the uniform growth and size of all the grains in the specimen.
Abnormal grain growth is when a few grains grow much larger than the remaining majority.
[17]
Grain boundary energy/tension
The atoms in the GB are normally in a higher energy state than their
equivalent in the bulk material. This is due to their more stretched
bonds, which gives rise to a GB tension

. This extra energy that the atoms possess is called the grain boundary energy,

.
The grain will want to minimize this extra energy thus striving to make
the grain boundary area smaller and this change requires energy.
[17]
“Or, in other words, a force has to be applied, in the plane of the
grain boundary and acting along a line in the grain-boundary area, in
order to extend the grain-boundary area in the direction of the force.
The force per unit length, i.e. tension/stress, along the line mentioned
is σGB. On the basis of this reasoning it would follow:

with dA as the increase of grain-boundary area per unit length along the line in the grain-boundary area considered.”
[17] [pg 478]
The GB tension can also be thought of as the attractive forces
between the atoms at the surface and the tension between these atoms is
due to the fact that there is a larger interatomic distance between them
at the surface compared to the bulk (i.e.
surface tension).
When the surface area becomes bigger the bonds stretch more and the GB
tension increases. To counteract this increase in tension there must be a
transport of atoms to the surface keeping the GB tension constant. This
diffusion of atoms accounts for the constant surface tension in
liquids. Then the argument,
holds true. For solids, on the other hand, diffusion of atoms to the
surface might not be sufficient and the surface tension can vary with an
increase in surface area.
[18]
For a solid, one can derive an expression for the change in Gibbs free
energy, dG, upon the change of GB area, dA. dG is given by
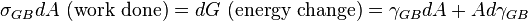
which gives
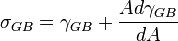
is normally expressed in units of

while
is normally expressed in units of


since they are different physical properties.
[17]
Mechanical equilibrium
In a two-dimensional
isotropic material
the grain boundary tension would be the same for the grains. This would
give angle of 120° at GB junction where three grains meet. This would
give the structure a
hexagonal pattern which is the
metastable state (or
mechanical equilibrium)
of the 2D specimen. A consequence of this is that to keep trying to be
as close to the equilibrium as possible. Grains with fewer sides than
six will bend the GB to try keep the 120° angle between each other. This
results in a curved boundary with its
curvature
towards itself. A grain with six sides will, as mentioned, have
straight boundaries while a grain with more than six sides will have
curved boundaries with its curvature away from itself. A grain with six
boundaries (i.e. hexagonal structure) are in a metastable state (i.e.
local equilibrium) within the 2D structure.
[17] In three dimensions structural details are similar but much more complex and the
metastable structure for a grain is a non-regular 14-sided
polyhedra
with doubly curved faces. In practice all arrays of grains are always
unstable and thus always grows until its prevented by a counterforce.
[19]
Grains strive to minimize their energy, and a curved boundary has a
higher energy than a straight boundary. This means that the grain
boundary will migrate towards the curvature.
[clarification needed]
The consequence of this is that grains with less than 6 sides will
decrease in size while grains with more than 6 sides will increase in
size.
[20]
Grain growth occurs due to motion of atoms across a grain boundary.
Convex surfaces have a higher chemical potential than concave surfaces
therefore grain boundaries will move toward their center of curvature.
As smaller particles tend to have a higher radius of curvature and this
results in smaller grains losing atoms to larger grains and shrinking.
This is a process called Ostwald ripening. Large grains grow at the
expense of small grains. Grain growth in a simple model is found to
follow:

Here
G is final average grain size,
G0 is the initial average grain size,
t is time,
m is a factor between 2 and 4, and
K is a factor given by:

Here
Q is the molar activation energy,
R is the ideal gas constant,
T is absolute temperature, and
K0 is a material dependent factor.
Reducing grain growth
- Solute ions
If a
dopant is added to the material (example: Nd in BaTiO
3)
the impurity will tend to stick to the grain boundaries. As the grain
boundary tries to move (as atoms jump from the convex to concave
surface) the change in concentration of the dopant at the grain boundary
will impose a drag on the boundary. The original concentration of
solute around the grain boundary will be asymmetrical in most cases. As
the grain boundary tries to move the concentration on the side opposite
of motion will have a higher concentration and therefore have a higher
chemical potential. This increased chemical potential will act as a
backforce to the original chemical potential gradient that is the reason
for grain boundary movement. This decrease in net chemical potential
will decrease the grain boundary velocity and therefore grain growth.
- Fine second phase particles
If particles of a second phase which are insoluble in the matrix
phase are added to the powder in the form of a much finer powder than
this will decrease grain boundary movement. When the grain boundary
tries to move past the inclusion diffusion of atoms from one grain to
the other will be hindered by the insoluble particle. Since it is
beneficial for particles to reside in the grain boundaries and they
exert a force in opposite direction compared to the grain boundary
migration. This effect is called the Zener effect after the man who
estimated this drag force to

where r is the radius of the particle and λ the interfacial energy of
the boundary if there are N particles per unit volume their volume
fraction f is

assuming they are randomly distributed. A boundary of unit area will
intersect all particles within a volume of 2r which is 2Nr particles. So
the number of particles n intersecting a unit area of grain boundary
is:

Now assuming that the grains only grow due to the influence of curvature, the driving force of growth is

where (for homogeneous grain structure) R approximates to the mean
diameter of the grains. With this the critical diameter that has to be
reached before the grains ceases to grow:

This can be reduced to

so the critical diameter of the grains is dependent of the size and volume fraction of the particles at the grain boundaries.
[21]
It has also been shown that small bubbles or cavities can act as inclusion
More complicated interactions which slow grain boundary motion
include interactions of the surface energies of the two grains and the
inclusion and are discussed in detail by C.S. Smith.
[22]
Natural sintering in geology
In
geology
a natural sintering occurs when a mineral spring brings about a
deposition of chemical sediment or crust, for example as of porous
silica.
[23]
A sinter is a mineral deposit that presents a porous or vesicular texture; its structure shows small cavities. These may be
siliceous deposits or
calcareous deposits.
[24]
Siliceous sinter is a deposit of
opaline or
amorphous silica which appears as incrustations near
hot springs and
geysers. It sometimes forms conical mounds, called geyser cones, but can also form as a
terrace. The main agents responsible for the deposition of siliceous sinter are
algae and other vegetation in the water. Altering of wall rocks can also form sinters near
fumaroles and in the deeper channels of
hot springs. Examples of siliceous sinter are
geyserite and
fiorite. They can be found in many places, including
Iceland, El Tatio geotthermal field in
Chile,
New Zealand, and
Yellowstone National Park and
Steamboat Springs in the USA.
Calcareous sinter is also called
tufa, calcareous tufa, or calc-tufa. It is a deposit of
calcium carbonate, as with
travertine.
Called petrifying springs, they are quite common in limestone
districts. Their calcareous waters deposit a sintery incrustation on
surrounding objects. The precipitation is assisted with mosses and other
vegetable structures, thus leaving cavities in the calcareous sinter
after they have decayed.
[24]
Petrifying spring at
Pamukkale,
Turkey:
Sintering of catalysts
Sintering is an important cause for loss of
catalyst
activity, especially on supported metal catalysts. It decreases the
surface area of the catalyst and changes the surface structure.
[25]
For a porous catalytic surface, the pores may collapse due to
sintering, resulting in loss of surface area. Sintering is in general an
irreversible process.
[26]
Small catalyst particles (which have the highest relative surface
areas) and a high reaction temperature are in general both factors that
increase the reactivity of a catalyst. However, these factors are also
the circumstances under which sintering is occurring.
[27] Specific materials may also increase the rate of sintering. On the other hand, by
alloying catalysts with other materials, sintering can be reduced. Especially
rare earth metals have been shown to reduce sintering of metal catalysts when alloyed.
[28]
For many
supported metal catalysts, sintering starts to become a significant effect at temperatures over 500 °C (932 °F).
[25] Catalysts that operate at higher temperatures, such as a
car catalyst,
use structural improvements to reduce or prevent sintering. These
improvements are in general in the form of a support made from an inert
and thermally stable material such as
silica,
carbon or
alumina.
[29]
See also
For the geological aspect :
References










No comments:
Post a Comment